Peripheral and Mucosal Immunity:
Critical Issues for Oral Vaccine Design
Arthur O. Anderson
INTRODUCTION
Promising new technologies from recombinant DNA and biodegradable polymer research will change how vaccines are produced and used (Michalek et al., 1995; Walker, 1994; Anderson, 1997). Two recent papers entitled, "Generation and Assembly of Secretory Antibodies in Plants" (Ma et al., 1995) and "Oral Immunization with a Recombinant Bacterial Antigen Produced in Transgenic Plants" (Haq et al., 1995) demonstrated that large-scale agricultural production of recombinant vaccine antigens and functional recombinant human antibodies is feasible and can yield quantities of product sufficient to satisfy virtually any requirement, including "edible" vaccines.
Recombinant vaccines formulated for oral use (i.e. encapsulated in biodegradable polymers) will provide multiple advantages over conventional vaccines (Morris et al., 1994; Michalek et al., 1994). Combining encapsulated vaccines with nutritious foods will be more convenient and acceptable to use, and will be easier to administer periodically. Oral vaccines will also save time and money. It will no longer be necessary for delays to assemble troops for immunization. Because the new vaccines can be self administered, they can be taken without the assistance of medically trained personnel, virtually eliminating issues surrounding the use of hypodermic needles. Taken together, it is now feasible to provide complete protective immunity along with good nutrition in "meals ready to eat" (Anderson, 1997). These new technologies should become an exciting active area of applied research, with numerous opportunities for interdisciplinary collaboration.
CURRENT CONCEPTS AND ISSUES IN IMMUNITY
It may be useful to digress from the primary objective (i.e., that of describing the oral vaccine technologies) to explain relevant concepts and critical issues in immunology. This explanation will help establish the importance of certain new approaches to immunization and reveal how these approaches could benefit soldiers of the 21st century. More thorough coverage of these topics may be found in the text: Mucosal Immunology
Compartmentalization of Immune Responses
Immune responses to vaccines are influenced by the route of immunization (injection or oral), the form of the antigen (live, killed, soluble, peptide subunit, particulate, etc.) and the presence in the vaccine of biologically active elements that mediate specific tissue tropisms. They may also contain adjuvants, vectors or vehicles (Walker, 1994; Spriggs, 1996) that effect the quality and quantity of immune response. For example, differences in immune responses to live vs inactivated virus vaccines may relate to antigen compartmentalization or to activities requiring vaccine viability like tropism or viral strategies for cell entry and replication (Rubin et al., 1986). In this review, general comments about effects of route or vehicles to immune responses will be restricted to those initiated by simple, unmodified, non-replicating, protein antigens to avoid confusion.
There is now substantial evidence supporting the existence of at least two immune systems, a "peripheral" immune system and a "mucosal" immune system (Ogra et al., 1994). These systems operate separately and simultaneously in most species including humans. Protective immunity acquired during convalescence is usually referred to as "systemic immunity," but this term is imprecise.
Systemic immunity might be a combination of mucosal and peripheral immunity or it might be dominated by an incomplete form of immunity dictated by a specific pathogen (Salgame et al., 1991; Mosmann and Sad, 1996). For example, if a pathogen stimulates a cytokine cascade that favors antibody production (at the expense of endocytosis and intralysosomal killing), it would continue to prosper within a compartment where antibodies are ineffective (Yamamura et al., 1991; Finkelman, 1995). Unless a vaccine stimulates the appropriate system or a combination of systems, immunity might not be complete.
The concept of anatomic compartmentalization of immunity is supported by observations from several disciplines (Kroemer et al., 1993). The anatomy of antigen uptake, physiology and biochemistry of lymphocyte recirculation, and unique tissue microenvironments, have functions that significantly influence the quality of humoral and cellular immune responses.
The way antigens are acquired by individual lymphatic tissues affects the outcome of an immune response. For example, the same antigen may produce qualitatively different immune responses in lymph nodes, spleen or Peyer's patches (Anderson, 1990). Antigens in lymph are filtered, trapped, processed and presented where the lymph passes over fixed antigen-presenting cells in lymph nodes. Such antigen handling by lymph nodes most often results in "peripheral immunity," characterized by the appearance of specific IgG in the blood. Antigens in blood are filtered, trapped, processed and presented in strategic blood/tissue interfaces in the spleen. This also results in "peripheral immunity." However, the spleen microenvironment is somewhat more complicated because it also accommodates circulating antigen-presenting cells and immunoreactive T- and B cells from other tissues committed to either peripheral or mucosal immunity. Antigens in the lumens of enteric organs (i.e., the respiratory and gastrointestinal tracts) are non-destructively endocytosed by specialized epithelial cells called "M" cells and transcytosed onto lymphoid cells in Peyer's patches where response to antigen presentation triggers commitment to "mucosal immunity" characterized by release of specific IgA into the secretions.
{kind=link}
Lymphocyte traffic patterns, regulated by selective expression of adhesion proteins in peripheral or mucosal lymphatic tissues, maintain anatomic segregation of immunological memory by causing antigen-primed cells to return to specific anatomic destinations where they will encounter conditions that further facilitate expression of peripheral or mucosal immunity (Kantele et. al, 1997). Among potentially myriad factors, these conditions include prevalence of specific cytokines, adhesion to- and costimulation by specific stromal cells, and still unknown microenvironmental factors intrinsic to those lymphoid compartments that favor commitment of B cells to specific immunoglobulin isotypes or T cells to peripheral or mucosal immunity.
Compartmentalized Humoral Immunity
All immunoglobulins participate in peripheral or mucosal immunity by binding antigen in a pocket formed by the complementarity-determining regions (CDR) encoded by the immunoglobulin heavy and light chain variable region genes (Carayannopoulos and Capra, 1993). The pocket formed by the VH/VL CDRs spatially conforms to the surface shape of the antigen with which the antibody binds.
The class of the antibody, conferred by the heavy chain constant (C) region genes, determines how it will function and where it will act. Antibody C-region genes are expressed after rearrangement of the selected heavy chain gene and assembly to the already rearranged variable, diversity and joining region genes. Thus IgM, IgD, IgG3, IgG1, IgG2a, IgG2b, IgE and IgA each have heavy chains that control how they participate in immunity, especially with regard to third-party molecular interactions such as Fc receptor binding, activation of the complement system and endosomal transport across mucosal epithelial cells.
Peripheral Immunity and IgG (Primarily IgG2a). Antibodies of peripheral immunity protect the parenchymal organs and peripheral anatomic sites bathed in tissue fluid and supplied by the blood microvasculature. These antibodies maximize cellular uptake and internalization of antigens. After pathogens have breached the barriers of the skin and/or mucous membranes, antibodies of the IgM and IgG subclasses work in conjunction with the complement system. This collaboration serves to neutralize, injure, aggregate and opsonize the pathogens so that they may be engulfed and destroyed by phagocytes.
Except in rare instances where pentameric IgM (complexed with J chain) may be secreted across epithelium, most circulating antibodies of the IgM and IgG subclasses work in blood, lymph and tissue fluids. They do not normally appear in mucosal secretions (Underdown and Mestecky, 1994).
Mucosal Immunity and Secretory IgA. Antibodies of mucosal immunity function outside the body at luminal surfaces of the moist epithelium lining conjunctiva, nasopharynx, oropharynx, gastrointestinal, respiratory and urogenital tracts and in the ducts or acini of exocrine glands. The principal antibody involved in mucosal immunity is secretory IgA (Underdown and Mestecky, 1994). This class of antibody requires the cooperation of two cell types for optimal activity in vivo. One cell makes the IgA and another cell transports it to the gut lumen where it works. The antibody-forming plasma cell releases dimeric IgA, postranslationally associated with J chain. The J chain holds the two IgA molecules together and facilitates binding to the poly-Ig receptor displayed on the abluminal side of epithelial cells. The complex is transported in endosomes to the luminal side of the epithelial cell and released into the secretions. The portion of the poly-Ig receptor retained with secreted IgA is called secretory component.
Pathogens adapted to infect mucosae express virulence factors that allow them to adhere, colonize or invade epithelium. Secretory IgA prevents absorption of these viruses, bacteria and toxins by blocking their adhesion while they are still on the external side of the epithelial barrier. This activity is opposite to that of antibodies associated with peripheral immunity. By preventing cellular attachment of the antigen, IgA enables it to be flushed away in the stream of secreted fluids and mucous washing over the epithelial membranes.
IgA may also facilitate transport of pathogens and toxins out of the body by causing them to be conveyed into bile and other exocrine secretions (Mazanec et al., 1993). Antigen-specific IgA has recently been shown to neutralize viral pathogens during transport across "M"cells of Peyers patches, where nondegradative endosomal transport might otherwise deliver a pathogen into the host (Owen and Jones, 1974; Neutra and Kraehenbuhl, 1994, Mazanec, 1992).
IgA is the preponderant antibody manufactured by the body. This escaped appreciation for many years because the blood contains a relatively low concentration of IgA compared to other immunoglobulins. However, 75 percent of the antibody-producing cells in the body make IgA, and most of this IgA is released continuously into gastrointestinal fluid, saliva, tears, urine and other secretions. Up to 40 mg/Kg body weight of IgA is manufactured and secreted daily in humans, which is many orders of magnitude greater than that of all other immunoglobulin isotypes (Brandtzaeg, 1994).
Compartmentalized Cell-Mediated Immunity
T lymphocytes involved in peripheral and mucosal cell-mediated immunity are known to segregate into functional subclasses of increasing diversity (Punt and Singer, 1996). T-helper cells (CD-4) and T-cytotoxic/suppressor cells (CD-8) may both assume immunoregulatory roles during immune responses, or they may differentiate into effector cells that exhibit segregated traffic patterns and functions (Salgame, 1991; Anderson and Shaw, 1996; Ebnet et al., 1996). The cytokine profiles secreted by T cells direct immunoreactive cell commitment to either peripheral or mucosal immune functions.
Peripheral T cells. T cells committed to peripheral immunity circulate in the blood, are involved in helping B- or T-cell precursors differentiate into antibody-secreting cells or cytotoxic effector cells. These T cells also release a menu of cytokines that activate and arm mononuclear phagocytes and natural killer cells to destroy intracellular parasites.
Mucosal T cells. Mucosally committed T cells enter Peyer's patches, the lamina propria and the intraepithelial compartment. There is considerable phenotypic diversity among these mucosal T cells. The subclass of mucosa-homing T cells, known as intraepithelial lymphocytes, are believed to be involved in protecting mucosal surfaces from injury by infectious pathogens or parasites (London et al., 1994). However, some of the diverse cells that populate the intraepithelial compartment include thymic independent T cells that enter the epithelium to complete maturation (Punt and Singer, 1996). Many of these cells display phenotypic markers that reflect intermediate stages of T-cell differentiation. (CD8+ IEL) The exact functions of IEL subpopulations are not yet known, but animal experiments and ex vivo cellular cytotoxicity assays suggest that some IEL exhibit cellular cytotoxicity directed against viral antigens on infected mucosal epithelial cells (London et al., 1987; Cuff et al., 1993). IEL are also thought to be involved in initiating tolerance (Elson et al., 1995; Gelfanov et al., 1996; Sim, 1995), but it is not clear whether all IEL, or a unique subset of IEL, induce tolerance to food antigens. Presentation of antigen by non-classical MHC antigens displayed on basolateral membranes of intestinal epithelial cells in the absence of costimulatory signals (Robey and Allison, 1995) may be involved in triggering tolerogenic T cells (Matzinger, 1994). There are many basic questions to be answered in the rapidly enlarging field devoted to IEL function.
Compartmentalized Immunity and Lymphocyte Traffic.
Immunity depends upon continuous movement of cells through blood, tissue and lymph (Anderson and Shaw, 1996). Lymphoid cells travel to the secondary lymphoid organs of the spleen, lymph nodes and Peyer's patches to encounter antigens acquired from the environment via blood, lymph or across mucous membranes. Where and by which cells antigens are presented to the trafficking cells has a significant influence on the outcome of the immune response with respect to antibody isotype commitment and future homing preference of memory and effector lymphoid cells.

Recirculation is the Integrating Principle. Recirculation of a precursor pool of uncommitted lymphocytes from the blood into lymph nodes or mucosal lymphatic tissues and then back to the blood again, forms the basis of immunosurveillance and integration of immune functions across the segregated systems. The magnitude of cell traffic reflected by the number of cells returned to the blood in efferent lymph is enormous. Enough lymphocytes recirculate from lymph to blood to replace the total blood lymphocyte pool from 10 to 48 times every 24 hours.
A consensus hypothesis explaining the overall process of emigration and mechanisms of organ-selective migration as a combinatorial mechanism has been proposed (Butcher, 1991). Circulating lymphocytes use adhesive selectins to roll on endothelial cell luminal surfaces; they become loosely tethered by lymphocyte L-selectin / sialyl Lewis-X interactions with endothelial cells. Upon activation by receptors expressed on the endothelial surface or by chemokines (chemotactic cytokines that emanate from between endothelial cells) the binding characteristics of these receptors are very rapidly (within milliseconds) changed from low affinity to high affinity (Anderson and Shaw, 1993; Ebnet et al., 1996). Different specific receptor-counter receptor interactions are responsible for each stage and mediate recognition, binding and emigration of cells from the blood.
Compartmentalized Traffic is Acquired. While naive lymphocytes appear to randomly access peripheral lymphoid tissues during recirculation, memory lymphocytes selectively return to the tissues where they first were stimulated by antigen. Precursor lymphocytes (that have not seen antigen in a lymphatic tissue or participated in an immune response) enter all tissues, especially secondary lymphatic tissues, such as Peyers patches, peripheral lymph node or spleen, without any organ selective bias. Shortly after activation by antigen, lymphocytes behave like inflammatory cells and avoid returning to secondary lymphoid tissues, preferring to lodge in skin, gut or inflammatory sites as "acute memory" cells. After maturation occurs, some of these memory cells will follow an organ-selective traffic route determined by the tissue in which that particular lymphocyte encountered the signals to divide differentiate and mature.
Conditions Influencing T-Cell Compartmentalization. Secondary lymphoid tissues draining peripheral or mucosal tissues differ from each other and therefore differently influence maturation of T cells (Cahill et al., 1977). If lymphocytes are activated in lymph nodes that drain skin, they become specialized to preferentially migrate into skin and serve the immunological needs of skin (Mackay et al., 1992; Mackay, 1991). Conversely, if lymphocytes are activated in lymphatic tissues, sampling antigens from the gut, they become specialized to preferentially migrate into the gut and serve its immunological needs (Kimpton et al., 1989; Abernethy et al., 1991).
Conditions Influencing B-Cell Compartmentalization. Primary B cells, expanding rapidly in primary mucosal lymphoid follicles of very young mammals, undergo diversifying mutations in the genes encoding the antigen-binding sites during VDJ rearrangement (Weinstein et al., 1994). They are then subject to positive selection to generate an antibody repertoire biased toward antigens prevalent in the environment; progeny that do not bind antigen in this milieu undergo apoptosis. Survivors leave the follicles in efferent lymph and enter the blood where they circulate and recirculate as small precursor IgM+/IgD+ B cells. They migrate continuously into all secondary lymphoid tissue until triggered to divide by the appropriate antigen and costimulatory signals.
Differentiation of circulating B cells depends on where they are first activated. If they first see antigen in peripheral lymphoid tissue (lymph nodes and spleen) they appear to be biased toward expression of antibodies associated with "peripheral" immunity. If the first see antigen in mucosal sites (such as Peyer's patches) they are more likely to be committed to IgA expression. The precise environmental stimuli that cause this commitment are not known but in vitro studies suggest that locally prevalent cytokines and unique stromal and accessory cell populations in the respective tissues are important (Weinstein and Cebra, 1991; Weinstein et al., 1991).
Some precursor B cells are first activated in spleen or lymph node. In that process, they are programmed to be one of the IgG isotypes (rather than IgA). Daughter cells from this clonal expansion leave the lymph node in the efferent lymph, return to the blood and seed the spleen, bone marrow, sites of inflammation and other lymph nodes as plasma cells and "memory" B lymphoblasts.
Other precursor B cells are first activated in Peyer's patches. Those cells become committed to rearrange their immunoglobulin heavy chain genes to express IgA (rather than IgG isotypes noted above); despite having made this commitment, the switch to IgA is often delayed for several days. This cell leaves in the efferent lymph, passes through the mesenteric lymph node (where it may be subject to immunoregulation by T cells it encounters there) and returns to the blood. These cells selectively migrate to the spleen, which serves as an auxiliary site for clonal expansion before dissemination of the daughter cells via the blood into mucosal lamina propria. This process takes 5-7 days, at least four trips in the blood and two to three cell generations (Cebra et al., 1977).
ISSUES FOR CONVENTIONAL MODES OF VACCINATION
Complicated Diseases Require Peripheral and Mucosal Protective Immunity
Protection from diseases that require complex forms of immunity require consideration of multiple "ancillary" factors. The nature of the vaccine antigen and route of its administration may confer anatomic compartmentalization to the immune response, restricting it to peripheral or mucosal immunity. Some infectious disease organisms spread parenterally to involve parenchymal organs but do not infect the epithelial cells lining respiratory, gastrointestinal or urogenital tracts. Protection against these pathogens depends on circulating IgM and IgG antibodies and/or peripheral cell-mediated immunity for resolution. Other disease agents principally infect mucosal epithelium, causing serious nutritional and pathophysiological alterations; but, in these diseases, circulating antibodies of the IgG class are not protective. Instead, secretory IgA and/or intraepithelial cytotoxic lymphocytes are needed for protection (Handbook of Mucosal Immunology, 1994).
Once A multitropic pathogen enters the body across the mucosae of the eyes, nose, throat, lungs and gastrointestinal tract, it might be able to disseminate and initiate systemic parenchymal as well as mucosal pathology. Parenteral immunization, the most common route of vaccination, usually elicits a peripheral immune response, with protective IgM/IgG antibodies and peripheral cell-mediated immunity. Parenteral immunization usually fails to stimulate mucosal lymphatic tissues to generate protective IgA antibodies or antigen-specific IEL. Many hazardous agents infect or intoxicate across the mucosae but spread through the systemic circulation. Protection against these agents requires vaccines that induce both peripheral and mucosal immune responses.
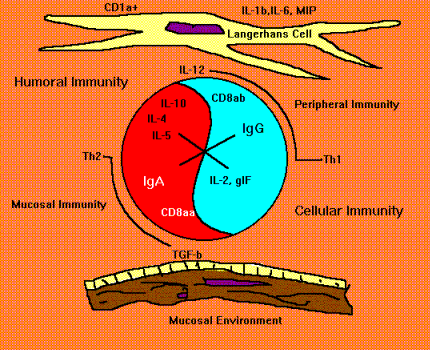
Cross Regulation is an Issue with Conventional Modes of Vaccination. Early studies suggested that the route of vaccination was crucial for determining whether protective peripheral or mucosal immunity would develop. Furthermore, immunization methods and routes for inducing mucosal immunity often delayed or prevented induction of peripheral immunity and vice versa (Chase, 1946; Mattingly and Waksman, 1978; MacDonald, 1982). This phenomenon, which I call "cross regulation" (Figure below), was first revealed in its bimodal form by Pierce and Koster (Pierce, 1978; Koster and Pierce, 1983) and Hamilton et al. (1979) when they were testing a non-toxic cholera vaccine candidate. They found that initial priming by vaccinating parenterally reduced the ability of the vaccine to elicit immunity in the mucosae when the second inoculation of antigen was given by an enteric route. But parenteral priming resulted in a strong booster response when the second dose of antigen was also given parenterally.

When Brown et al. (1981, and unpublished) empirically tested the effect of route of immunization of a Formalin inactivated Rift Valley fever virus (RVF) vaccine on protection from parenteral or mucosal challenge, he found that the nonreplicating vaccine elicited an immune response that protected mice from parenteral challenge but not from aerosol challenge. This asymmetric immunity was also found for the C-84 Venezuelan equine encephalitis virus vaccine (Jahrling and Stephenson, 1981). We repeated these experiments using a matrix design that tested both routes of immunization against homologous and heterologous challenge routes (Anderson et al., 1988; Hart et al., 1995; Pitt and Anderson, unpublished). Upon heterologous challenge, viral pathogenesis (target organs affected and time course of disease evolution) was altered. Protection from encephalitis was deficient when subcutaneously immunized mice were challenged by aerosol. And, when mucosally immunized mice were challenged subcutaneously, the onset of viral infection of the liver extended over 18 days. Usually, the liver was affected only during the first 3-6 days. The levels of specific IgA or IgG in serum and secretions corresponded to whether the challenged animals experienced protection and/or altered pathogenesis (Anderson et al., 1988).
Oral Tolerance and Mucosal Immunity. There is no easy way to explain this phenomenon. The mucosal immune system is known to be the site of priming for two paradoxically opposite purposes, i.e., tolerance and mucosal immunity. The usual response of the gastrointestinal tract to antigens is tolerance rather than immunity (Chen et al., 1995). The mechanisms of oral tolerance remain unclear. Tolerance could be primed in Peyer's patches or in the intraepithelial compartment. I think IEL are good candidates for communicating a toleragenic signal. Epithelial cells can present antigens; but, without important costimulatory molecules, the cells that see this antigen may die or be rendered non-responsive (Matzinger, 1994). It is thought that tolerance can occur through active suppression (cells or cytokines that induce non-responsiveness), anergy (live, non-responsive cells), or apoptosis (signal programs cell death) of antigen-reactive cells.
Tolerance versus Cross Regulation. The healthy gastrointestinal tract is bathed in an enormously diverse collection of environmental antigens, yet only certain antigens stimulate active mucosal, peripheral or combined mucosal and peripheral immune responses. Somehow the tissue is able to distinguish pathogens (dangerous) from normal flora (safe) and food antigens (safe) (Matzinger, 1994). Inducible chemokines may be the mucosal "danger" signal (Oppenheim, et al., 1991; Eckmann et al., 1993). Pathogens that bind and/or invade mucosal epithelium cause epithelial cells to release cytokines and chemokines that attract inflammatory and/or immune cells, or cause epithelial cells to express proteins (classical and non-classical MHC) that target antigens for induction of immunity (Eckmann et al., 1993; Agace et al., 1993; and McCormick et al., 1993). Mucosal antigens that lack costimulatory activity are programmed for tolerance (Kagnoff, 1996; Bendelac, 1995). When tolerance results from mucosal immunization, it does not appear to diminish the stimulation of B cells committed to IgA secretion, however. Mucosal tolerance (Mowat, 1994) is most likely responsible for preventing systemic responses to common food antigens, but it has different conditions of triggering than that for cross regulation.
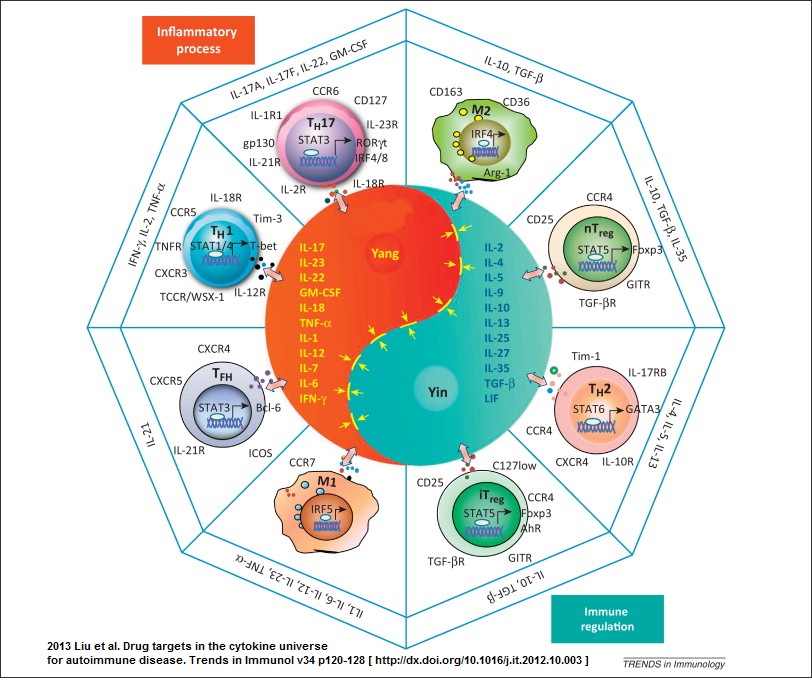
Caption: This diagram, resembling the oriental symbol for yin and yang, represents microenvironmental factors that stimulate T-cells to express either T-helper 1(Th1) or T-helper 2 (Th2) cytokines which are associated with "cellular" and "humoral" immunity, respectively. Other allignments of cytokines may contribute to cross-regulation of immune responses associated with parenteral or mucosal immunization. The cytokines IL-12 and TGF beta 1 are predominant influences in "peripheral" and "mucosal" lymphatic tissues. Thus vectorial expression of these cytokines affect T cells and B cells in such a way that proliferating B cells become committed to secrete "peripheral" IgG or "mucosal" IgA, respectively. Recent data also suggest that heterogeneity among dendritic cell populations may effect the balance of Th1/Th2 immunity. The complexity of this Yin Yang concept has increased as new information which added the dichotomy between inflammation and immunity to existing dichotomies of cellular vs humoral and peripheral vs mucosal immunity (this image). And, for the Inflammation vs Immunity dichotomy click on the yin yang image (above) and also see: Liu et al. Trends in Immunol v34 p120-128, 2012
Suppression. What some regard as mucosal or peripheral tolerance could merely represent a temporary induction of antigen-specific suppression. The suppression of systemic delayed cutaneous hypersensitivity and reduced IgG expression induced by mucosal priming lasts less than 3 months. In contrast true tolerance persists until some stimulus breaks its effect. We found that peripheral priming with antigens in peripheral lymph nodes resulted in reduced mucosal immune responses comparable to that mentioned above (Anderson et al., 1991; Anderson et al, 1988). This "yin yang" effect on peripheral versus mucosal immunity suggests that cross regulation might not be tolerance but indirectly be influenced by T helper cells secreting cytokines that exert positive or negative feedback control. This suppression may require cell contact or it may exert its effect through local cytokine commitment that prevents T-cell help of the required variety, i.e., you need IL-4 but you get IF gamma. This difference is interpreted as suppression.
There is a dichotomy of immune systems responding to vaccine antigens. Each immune system has its own stereotypical way of responding depending upon whether antigen gets there by parenteral or oral routes of immunization. The preponderant antibody type and effector T-cell type are shown. Each system has the ability to suppress the other by numerous mechanisms. Delayed hypersensitivity, regulated by preponderant Th1/Th2 cytokines, is not displayed in healthy mucosal tissues.
{kind=link}
T-helper Subclasses and Cross Regulation. Because cross regulation may influence immunity during the short term after oral or parenteral immunization, it is of value to discuss the possible role of T-helper cell subclasses in cross regulation (Street and Mosmann, 1991; Swain et al., 1991; Trinchieri, 1993). Helper T cells segregate into two subsets depending on whether the cells secrete interferon gamma (IFgamma and interleukin-2 (IL-2) and IL-12 (T helper-1, Th1) or IL-4. IL-5 IL-6 and IL-10 (T helper-2, Th2) (Mosmann and Sad, 1996). This distinction is important because the cytokines determine the kind of help provided by the respective T-helper cell types. This dualistic system that achieves harmony between opposing cytokine influences is graphically illustrated in Figure 6. The central object in figure 22-6 which resembles the symbol for yin and yang represents the symmetric opposing effects of cytokines on lymphatic tissue microenvironments. In "peripheral" lymphoid environments such as in lymph nodes draining the skin, IL-12 secreted by Langerhans cells favors differentiation of T cells that secrete IFN-gamma and IL-2. In addition to favoring "cellular immunity" these The cytokines favor production of IgG isotopes by B-cells. The "mucosal" environment in the Peyer's patch is affected by high local concentrations of TGFbeta1. This cytokine favors development of T cells that secrete IL-4, IL-5, IL-6, IL-10 etc. IL-4 negatively effects "peripheral cellular immunity" at the same time that it favors development of B-cells committed to IgA secretion. Furthermore, TGFbeta1 has been shown to directly affect immunoglobulin gene class switching to favor selection of "alpha" heavy chain genes. Thus, environmental influences, of which cytokines are a prominent few, affect the "direction" of the immune response toward "cellular" (Th1) or "humoral" immunity (Th2). When the vector is oriented alone the extreme opposites. However, intermediate "vectorial" orientations productive of mixed secretion of cytokines can produce "mucosal" or "peripheral" immunity when IL-4 and IFN gamma moderately predominate among other microenvironmental cues.
The relative concentration of T-helper types in lymph nodes or Peyer's patches influence selective expression of IgG or IgA immunoglobulin isotypes (Stavnezer, 1995). Anatomic compartmentalization of T-helper subclasses is of value when considering possible mechanisms of cross regulation. When Th1 responses are preponderant (as they are in skin-draining lymph nodes), T helper cells secrete IL 2, IL-12 and IF gamma, resulting in selective expression of IgG immunoglobulins and activation of cytotoxic T cells and armed mononuclear phagocytes (Weinstein et al., 1991; Kang et al., 1996; and Ariizumi et al., 1995). Where Th2 responses are preponderant (as they are in mucosal sites), T helper cells secrete IL 4, 5, 6, 10, etc, resulting in selective expression of different immunoglobulin isotypes including IgA (Hiroi et al., 1995; Lebman and Coffman, 1994). T helper cells also down regulate peripheral cell-mediated immunity at the same time as there is increased development of cytotoxic T cells that selectively home to mucosal intraepithelial sites (Ke and Kapp, 1996). The T-helper cell types provide positive and negative feedback control on commitment of B cells to express specific antibody isotypes. For example, IF gamma suppresses commitment to IgA and IL-4 suppresses commitment to IgG. Whether this is a direct effect on the B cell or an indirect effect mediated by T cells is a subject of great interest.
Anatomic Compartmentalization of T-helper Subclass Commitment. In mucosal sites, abundance of the cytokine TGF beta-1 programs Th0 cells to develop into Th2 cells (Lebman and Coffman, 1994; Young et al., 1994). The cytokines secreted by Th2 cells contribute to expansion and differentiation of B cells committed to IgA expression. TGF beta-1 also contributes to selective expression of IgA antibodies by favoring immunoglobulin heavy chain gene switching to IgA, and by suppressing expression of other isotypes (Lebman and Coffman, 1994; Stavnezer, 1995). TGF beta-1 is not widely distributed in peripheral lymph nodes where there is selective expression of Th1 cellular responses and IgG antibodies. Recent publications suggest that Langerhans cells from the periphery may have a role in Th1 commitment by secreting IL-12 and IL-1 beta - converting enzyme (Kang et al., 1996; Ariizumi et al., 1995). Other relationships of Th1/Th2 responses may depend upon which antigen-presenting cell type migrates into a site and conditions the microenvironment (Morikawa et al., 1995).
When this system is analyzed in vitro at the single cell level, specific Th types or cytokine profiles are not sufficient to cause uncommitted IgM+, IgD+ or IgM-only B cells to switch to IgA (Shrader et al., 1990, Kotloff and Cebra, 1988). However, when antigen-presenting cell types or stromal cells are added, switching occurs. Thus, the cross regulation is related to a sum of multiple "microenvironmental" factors unique to the tissue where the immune response is initiated (Weinstein and Cebra, 1991, Weinstein et al., 1991). Resolution of this on a molecular level awaits further research study.
Cross Regulation is Incompatible with Complete Protective Immunity. Cross regulation is not a desirable feature of conventional modes of vaccination. We and others urgently began looking for effective ways to overcome the cross regulation that prevented simultaneous induction of mucosal and peripheral immunity. Not all vaccine antigens exhibit this behavior, especially those that have so-called "adjuvant" effects. We found that modifying the vaccines with immunological adjuvants was a partial solution, but the adjuvants had to be used at optimal doses that were different for each vaccine tested (Anderson, 1985; Anderson and Reynolds, 1979; Anderson and Rubin, 1985; Anderson et al., 1987). Adjuvants were not effective with all antigens and empiric studies had to be performed to select the optimal combinations. We observed that certain adjuvants (such as the lipoidal amine Avridine) also potentiated the undesirable cross regulation observed with native antigen (Anderson et al., 1985; Anderson et al., 1987). For example, if an antigen given orally elicits a good IgA response in secretions but fails to elicit or slightly depresses specific IgG in serum, use of an adjuvant with the antigen would increase the IgA response and further decrease or block the IgG response in serum.
SOLUTIONS TO CROSS REGULATION FROM NEW TECHNOLOGIES
When our approach was defining cross regulation, other scientists were finding that they could induce simultaneous peripheral and mucosal immunity. They were working with biodegradable microspheres and enterotoxin-derived, vaccine-delivery systems. Antigens administered orally in biodegradable microspheres composed of poly (lactide/glycolide) copolymer, or antigens associated with carrier proteins derived from cholera toxin (CT), or Escherichia coli heat-labile enterotoxin (LT), induced simultaneous peripheral and mucosal immunity (Eldridge et al., 1989; Elson and Ealding, 1984a,b). The same or similar antigens given without these particular vehicles yielded cross regulation. In addition, antigens conjugated with the B-subunits of CT or LT triggered acceptable peripheral and mucosal immune responses that were unattainable when each antigen was given alone (Dertzbaugh and Elson, 1991, Holmgren et al., 1993; and Elson and Dertzbaugh, 1994). These results provided evidence that cross regulation could be overcome by using non-toxic and biodegradable vaccine vehicles.
Solutions from Microencapsulation Technology
The first technology I wish to highlight is microencapsulation of vaccines with biodegradable polymers for immunization of the peripheral and mucosal immune systems. There are a number of recent reviews (there are even new journals committed to the field) that cover all the new microencapsulation systems, polymer chemistry (Gombotz and Pettit, 1995; Langer, 1990) and biological applications (Walker, 1994; Michalek et al., 1994; Morris et al., 1994; ). After reports of success in enhancing immune responses with adjuvant and particulate antigens, numerous particulates were tested for vaccine enhancing activities. The chemical composition of particles that were tested included polystyrene, latex, poly(methylmethacrylate), polyacrylamide, poly(butyl-2-cyanoacrylate), alginate, ethyline-vinyl acetate copolymer, and poly(lactide glycolide) copolymer (Michalek et al., 1994). These polymers exhibit a broad range of utility, toxicity and biodegradability. I will restrict my discussion to observations about the use of polymers comprised of lactic and glycolic acids (natural products of energy metabolism) as vehicles because they have been tested for efficacy in initiating peripheral and mucosal immune responses. Poly (lactide/glycolide) copolymer was originally developed for biodegradable suture material (Morris et al., 1994). It is licensed by the FDA for that use and has been used for many years in human subjects with no serious adverse sequelae. During the past 10 years, there has been exponential development of microencapsulation technology for the delivery of drugs and hormones in addition to other uses such as the scratch-and-sniff advertisements found in magazines.
Biodegradable Microspheres have Adjuvant Effects. In the early 1980's, the technology was first successfully applied to parenteral and oral immunization with vaccines by Dr. John Eldridge. In addition to demonstrating that encapsulation protected the antigen from deterioration or degradation in the gastrointestinal tract, Dr. Eldridge revealed that microencapsulation provided an "adjuvant" effect. The same quantity of antigen in microcapsules elicited more vigorous immune responses than antigen given alone.
Many of his vaccine formulations were inoculated parenterally to determine antigen-release kinetics. The rate of particle degradation (conferred by molecular weight, surface area and ratio of lactide to glycolide in polymer composition) could be utilized effectively to control the rate of antigen release. It was discovered that microcapsule formulations could be constructed which released antigens in pulses that mimicked a multiple inoculation schedule, although the product was administered only once. This line of inquiry has prospered and new improved formulations have been developed that facilitate encapsulation of aqueous phase antigens and various water-in-oil or water-in-oil-in-water emulsions (Yan et al., 1994).
Microencapsulated Vaccines overcome Cross Regulation. When vaccine antigen in poly (lactide/glycolide) copolymer was administered orally in heterogeneous sizes, including 10 micrometer and >1 micrometer sizes, particles of >10 micrometer diameter were not absorbed in the gut. The larger absorbable particles (diameters >5 to ~10 micrometers) arrested in Peyer's patches, whereas the smaller particles (<5 to ~1 micrometers) were disseminated to mesenteric lymph nodes and spleen (Eldridge et al., 1990). This bimodal anatomic distribution of particles was accompanied by simultaneous elicitation of IgG and IgA immune responses. Hydrophobicity enhanced uptake of the particles in Peyer's patches and hydrophilicity favored dissemination. The size of the particles appeared to be the critical determining factor that enabled simultaneous induction of peripheral and mucosal immune responses.
Solutions from Enterotoxin-derived Vaccine Carriers
Cholera toxin (CT) has important pharmacological effects. This has been known since it was determined that the watery diarrhea of cholera was not caused by intestinal epithelial cell injury (Gangerosa et al., 1960). The prodigious fluid output was produced by a pharmacological effect of CT on intestinal epithelial cells. After CT binds monosialyl GM1 ganglioside on epithelial cells via the pentameric B-subunit, the toxigenic A-subunit enters the cell and ADP-ribosylates the adenylate cyclase regulatory protein Gs. The result is run-away adenylate cyclase activity, which causes the absorptive epithelial cells to secrete massive amounts of fluid (Holmgren, 1981; Spangler, 1992).
Adjuvant Activity of Cholera Toxin. Cholera toxin is a potent immunogen given by any route (Pierce and Gowans, 1975; Fuhrman and Cebra, 1981). It is one of few antigens that will yield a potent IgA immune response when administered orally to experimental animals. Cholera toxin never induces tolerance or cross regulation in vivo. Furthermore, it can prevent a tolerogenic antigen from inducing tolerance when the two are given together. Cholera toxin may lose its ability to increase immunogenicity of nominal antigens if its B-subunit (CTB) is denatured or blocked, adversely affecting the GM1 ganglioside-binding capacity essential for adjuvanticity (Sun et al., 1994; Dertzbaugh and Elson, 1993). Conjugating antigens to CT or CTB can cause relatively poor mucosal immunogens to strongly stimulate peripheral and mucosal immune responses (Elson and Ealding, 1984 a,B; Dertzbaugh and Elson, 1991,1992; Holmgren et al., 1993; Elson and Dertzbaugh, 1994).
The adjuvant activity of CT can be obtained if it is mixed with antigen. On the other hand, non-toxic CTB must be linked to an antigen to produce an adjuvant effect. Sometimes the procedure used to chemically link antigens to CTB damages the tertiary structure of the pentameric B-subunit ring and reduces adjuvanticity. An objective of Dr. Dertzbaugh and our laboratory to develop a bacterial expression system that will genetically fuse an antigenic construct to CTB in such a way that it can be expressed without alterating the native configuration of the antigen or the B-subunit. A similar approach has already been reported with some success (Wu and Russell, 1994; Hajishengallis et al., 1995). Dr. Dertzbaugh and our group have been focusing on the benefits of using the non-toxic B-subunit (with or without molecular spacers derived from the non-toxic A2 peptide). Using constructs that lack the A1 ADP-ribosylase will avoid our having to deal with issues related to toxicity.
Immunohistochemistry (Anderson and Dertzbaugh, unpublished) reveals that some of the increased antibody production found when CT is used as an adjuvant may result from facilitated secretion by a few plasma cells rather than more cells secreting antibody. I have observed that CT induces B cells to secrete antibody when they are still in the intermediate stages of migration and differentiation. Normally, IgM- or IgG-secreting cells are infrequently seen in gut lamina propria. During the first 24 hours after oral CT treatment, IgM+, IgG+ and IgA+ cells accumulate antibodies in surrounding lamina propria indicating increased secretion. These effects of cholera holotoxin may be pharmacological rather than immunological because CT may cause plasma cells to hypersecrete by similar mechanisms as CT uses to cause diarrhea through epithelial hypersecretion.
CT and E. coli Heat-Labile Enterotoxin. Cholera toxin and LT of E. coli are very homologous in amino acid sequence (CT and LT have 80% amino acid homology, Dallas and Falkow, 1980) and tertiary structure (Sixma et al., 1991; Zhang et al., 1995; Burnette, 1994). LT is believed to have less potent toxicity than CT, but both cause diarrhea in human subjects at relatively low doses. The LT-1 enterotoxin has been used in similar adjuvant studies and found to have virtually identical effects as CT (Clements et al., 1988).
One critical difference between CT and LT is that the A subunit of CT is cleaved postranslationally into an enzymatic A1 and a non-toxic A2 peptide, whereas the A1 and A2 of LT must be cleaved by proteases encountered in the environment for toxicity to result. A report by Lycke et al. (1992) indicated that mutations that destroy the ADP-ribosylating activities of CT or LT also destroy the adjuvanticity. However, Dickinson and Clements (1995) were able to dissociate adjuvanticity from toxicity by creating a mutant resistant to proteolytic activation. The R192G mutant LT has an arginine substituted for a glycine at the 192nd amino acid from the N terminus, rendering the site resistant to proteolysis. This mutant LT (mLT) retains its ability to function as an adjuvant when given orally. Furthermore, its adjuvant activity extends to potentiating antigen-specific IgG serum and IgA mucosal antibody responses in mice (Dickinson and Clements, 1995). It also prevented induction of tolerance (cross regulation) to nominal antigens. This promising product is rapidly moving through pre-clinical and clinical safety and efficacy trials (personal communication, Commander Dan Scott and Dr. John Clements).
Effects of CT/LT on Mucosal T-Cell Compartments
The mechanisms that enable CT or LT to overcome cross regulation, prevent tolerance and potentiate peripheral and mucosal immune responses are not known. In vitro and in vivo studies of the cells, cytokines and tissues affected by CT or LT have not revealed many similarities to effects of traditional adjuvants like complete Freund's adjuvant (Freund, 1956; Anderson et al., 1987; Anderson, 1985). Perhaps CT and LT are merely improving the natural immune processes described earlier in this chapter, such as antigen presentation, cytokine secretion, lymphocyte traffic, etc.
Effect of CT or CTB on Lymphoid Tissue Compartments. T- and B-cell compartments in lymphatic tissues from mice that had been orally dosed with CT or CTB were examined by immunohistochemistry. There were no glaring changes in T- and B-cell populations in lymph nodes, spleen or Peyer's patches. However, I noticed that CT and CTB very quickly depleted the CD8+ cells in the intraepithelial compartment. These IEL are normally very prevalent. The rapid reduction by 80% of CD8+ IEL between 12 and 72 hours was impressive. Equally impressive was the finding that between 10 and 14 days later, the intraepithelial compartment was repopulated. Depletion of IEL primarily affected CD8+ cells, but other phenotypes must also have left the epithelium. The number of CD4+ cells in Peyer's patches and lamina propria stayed the same or increased during the drop in CD8+ cells. The depletion of IEL was triggered by doses of CT or CTB that previously were shown (in other studies) to be mucosal adjuvants (Elson and Ealding, 1984a,b). These anatomic findings were consistent with a hypothesis that CT/LT achieves its adjuvant effect by altering the T-cell regulatory environment in mucosal lymphoid compartments to support mucosal immunity, prevent tolerance induction and ameliorate cross regulation (Elson et al., 1995; Xu-Amano et. al., 1993). Effects of cholera toxin on microenvironment controlling cytokines such as IL-12 and/or TGFbeta could be involved in affecting the balance of Th1/Th2 immune responses. Indeed recent data suggest that cholera toxin may downregulate IL-12 expression, thereby removing influences that diminish committment to antibody production in general and to IgA, specifically.
Solutions to Quantity and Cost Issues for CT/LT Vaccine Carriers
The idea of developing new vaccines incorporating recombinant antigens and/or biologically active carriers raises a few questions. One concern is about yield. Bacteria reproduce rapidly but it takes a lot of bacteria a long time to produce only a handfull of product, and the cost of operating large fermenters is not trivial. Contamination of product with bacterial endotoxin may also affect the yield. The product must be purified of endotoxin which will reduce the total yield.
Another of the factors that might limit interest in developing recombinant vaccines and therapeutic antibodies is cost. It has often been stated that vaccines take 10 years or more from discovery to FDA licensure, if you are lucky. Many people start to develop vaccines, but few vaccines become licensed because anywhere along the way the product may fail. It will save money if it fails during the pre-clinical phase. Many other vaccines fail during Phase I safety trials in human volunteers if adverse reactions are unacceptable. The worst case would be failure during field tests when a vaccine, which was safe and efficacious in experimental animals, fails to corroborate its efficacy in humans.
Pharmaceutical industries face these realities all the time, but the public (our customers) can't understand why every start does not necessarily translate into a new beneficial product for them. Vaccine research and production cost time, personal commitment and lots of money. This becomes an especially frustrating concept if one considers that the vaccines needed most, such as malaria vaccines, are effective against disease threats that are endemic in countries in which the financial resources preclude any chance of buying the product.
The next section will show how both of these issues may be solved by producing antigens or antibodies in plants.
SOLUTIONS FROM PRODUCTION IN TRANSGENIC PLANTS
Rapid progress in plant biotechnology will result in contributions to vaccine production. Recent results suggest that genetically engineered plants may be used to produce vaccines against human diseases (Haq et al., 1995; Mason et al., 1994; and, Arntzen et al., 1994a,b). A concern is that plant-produced vaccines will have to be purified of alkaloids and other toxic plant materials. Two approaches may be used to resolve this. One is to use plant vectors that incorporate the protein into a tissue that lacks toxic alkyloids, e.g., bananas or soybeans (Arntzen et al., 1994b; McCabe et al., 1988). Another solution would be to use the same technology employed to incorporate the vaccine construct into plants to genetically remove the genes for the toxic substances (Dr. Mich Hein, personal communication).
If the potential concerns about plant alkaloids can be quickly resolved, production of vaccines in plants will be a significant improvement over current methods. Such vaccines might be cheaper than those now available because plants are easier to grow in large quantities than are cultured animal, insect, bacterial or yeast cells now used to make most vaccines.
The details of inserting antigenic constructs into plants as expression vectors are similar to those for baculoviruses, bacteria, or yeast. The discovery that plant tumors (called galls) are produced by a bacterium called Agraobacterium tumifasciens is responsible for this rapid progress in developing plants as expression vectors (Horsch et al., 1989); Jefferson et al., 1987). In addition, new technologies for inserting genes into plants have been developed to overcome limited host-range specificity of Agrobacterium vectors (McCaabe et al., 1988; Owens et al., 1985; Pedersen et al., 1983).
The plant biotechnology field has been defining the requirements for inserting and expressing genes in plants since the late 1970s. The current breakthrough has been spurred by the demonstration of efficient production of heterologous proteins in plants (Hiatt et al., 1989; Gasser and Fraley, 1989). This may produce a potential benefit for tobacco farmers because the easiest plant vector to transform is the tobacco plant. As more people stop smoking, the new technology might provide important replacement crops for tobacco growers if the toxic alkaloids in tobacco leaf can easily be removed by genetic engineering.
New biotechnology companies are well positioned to benefit from the anticipated economic benefits because all that would be needed to scale-up production of plant-made protein to the amounts needed for a commercial vaccine is to add acreage [FDA Guidance]. Cheaper vaccines that are available in large quantities make vaccination of an entire expeditionary force feasible in contingency situations. It is anticipated that using transgenic plants from one growing season as expression vectors might provide all the protein needed compared to conventional vaccine production techniques, which require much more time. These vaccines potentially will be safer because production in plants virtually eliminates contamination with animal viruses.
Charles Arntzen is developing "edible vaccines." These vaccines are designed with the expectation that no purification step will be required. The vaccine components are expressed in final form in the interstices of the plant. The use of a CTB or LT vaccine carrier would target the antigen construct to be taken up and processed in mucosal lymphatic tissues where the biological effects of the carrier should program the response for peripheral and mucosal immunity.
CONCLUSIONS
Mucosal immunology has contributed to the understanding of concepts and issues of host resistance needed for protection of the gastrointestinal tract and other mucosa that provide for optimal nutrition. An added benefit of support for this discipline has been a better understanding of how vaccines may be used more effectively.
The new technologies of vaccine microencapsulation, use of the non-toxic B-subunit of cholera or E. coli enterotoxin carriers for recombinant vaccine constructs, and incorporation of recombinant vaccine constructs in edible plants have become feasible. This technology can be studied for its acceptability for delivery of vaccines to military personnel.
The above technologies overcome cross regulation and induce immune responses with circulating IgG as well as secretory IgA which should provide complete active prophylaxis without need for needle injections or time-consuming immunization schedules.
Plant Geneticist Pamela Ronald Presents the case for engineering our food
REFERENCES
Click on "REFERENCES" above and they will appear
Scientists at UAB also feel mucosal vaccines will be useful in controlling "Emerging Infectious Diseases".
Read comments Art Anderson posted to NPR on how the immune system can cause chronic inflammatory disease as it responds to microbial antigens, and how the immune system takes cues from the neuroendocrine systems.
